


ADMIN | 2020-02-16 15:08:23
为推动我国生物信息学的学科发展和创新研究,充分展示和宣传我国生物信息学领域的重大研究成果,《基因组蛋白质组与生物信息学报》(Genomics, Proteomics &Bioinformatics, 简称GPB)于2018年首次组织评选了年度“中国生物信息学十大进展”,得到了广泛认可。在此基础上,2019年度中国生物信息学进展评选工作增设了分类评选,由领域同行专家首先评选出了2019年度“中国生物信息学十大数据库”、“中国生物信息学十大算法和工具”、“中国生物信息学十大应用”;并在分类评选的入选工作中进一步评选,产生2019年度“中国生物信息学十大进展”。
现公布2019年度“中国生物信息学十大进展”(排名不分先后,按题目首字母顺序排序),之前已公布2019年度“中国生物信息学十大数据库”、 “中国生物信息学十大算法和工具”及“中国生物信息学十大应用”的评选结果,欢迎一并关注!
感谢所有专家秉持专业和公正的态度参与本年度十大系列工作的推荐和评选,祝各位老师在新的一年取得更多优秀成果!同时,感谢我所BIGD冯昶瑞同学在本次活动资料整理和图片设计工作中所付出的努力!
发展创新助生信、众志成城战疫情。金鼠之年伊始,GPB祝愿大家平安健康,让我们在各自的岗位上做好自己的工作,为国民健康和国家发展贡献力量。Be a better me, be a better you, for a better us。中国加油!
GPB
2020年2月17日

利用单分子测序构建高质量基因组的算法—HERA
高质量基因组序列对于研究一个物种基因组的结构、功能、进化、基因定位和克隆等都至关重要。复杂基因组中存在大量的重复序列,无法用现有的基因组组装方法获得,严重影响了参考基因组的质量及其应用。中国科学院遗传与发育生物学研究所梁承志团队开发了利用单分子长片段测序,对基因组复杂重复序列区域进行高效组装的算法HERA。测试发现,HERA能够准确组装水稻中包括复杂的长串联重复序列在内的绝大部分重复序列。与相应物种已发表的基因组版本比较表明,利用HERA组装的玉米和人基因组的contig N50分别从1.3 Mb和8.3 Mb提升至61.2 Mb和54.4 Mb;新组装的苦荞基因组contig N50达到了27.85 Mb。新的玉米基因组组装版本在玉米B73参考基因组中填补了大量以前没有组装出的序列,对多处染色体上序列位置或方向的错误进行了校正,并增补了一些以前遗漏的多个重要基因序列。利用HERA改进后的苦荞全基因组8条染色体仅由20个contig组成,其中一条染色体仅包含一个contig。本工作展示了利用现有常规技术条件构建几乎完整的基因组的潜力。
该成果发表于《自然通讯》期刊。
工具链接:
https://github.com/liangclab/HERA
原文信息:
DuH, Liang C. Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads. Nat Commun 2019;10:5360. PMID: 31767853.
原文链接:
https://doi.org/10.1038/s41467-019-13355-3

图:HERA改进的玉米基因组与已发表的参考基因组B73 RefGen_v4的比较。(a)全基因组中序列缺口由2523个减少到了76个;(b)玉米参考基因组中缺失或多余的序列(上)经HERA改进后(下)被正确地填补或移除。
人类和小鼠细胞标志物数据库—CellMarker
飞速发展的单细胞测序技术为探究复杂疾病开辟了新道路。单细胞研究面临的首要问题是如何确定组织细胞类型,然而,目前尚缺乏一个可供参考查询的综合细胞标志物数据库。哈尔滨医科大学李霞、肖云团队和哈尔滨医科大学附属第一医院赵婷婷通过查询逾10万篇已发表的文献,构建了人类和小鼠组织中各种细胞类型的标志物数据库—CellMarker。该数据库囊括了158种人类组织中467种细胞类型的13,605个细胞标志物,以及81种小鼠组织中389种细胞类型的9148个细胞标志物,涉及131种癌症细胞标志物。CellMarker数据库存储的细胞标志物对细胞身份的识别和刻画提供了重要分析依据,对从单细胞水平解析疾病发生及微环境影响提供了帮助。
该成果发表于《核酸研究》期刊。
数据库链接:
http://biocc.hrbmu.edu.cn/CellMarker/
原文信息:
Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 2019;47:D721–8. PMID: 30289549.
原文链接:
https://doi.org/10.1093/nar/gky900
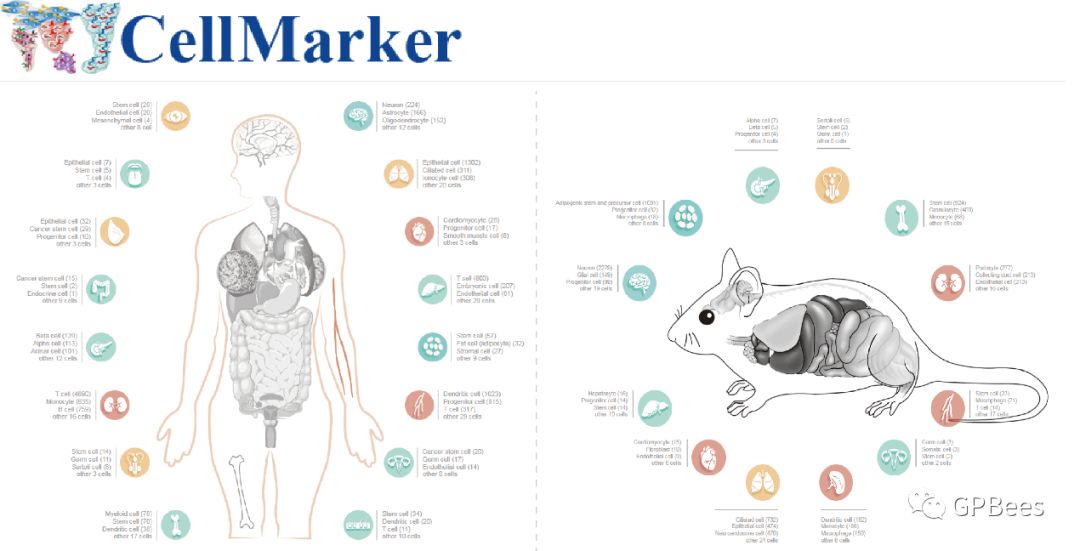
图:CellMarker数据库中人类和小鼠组织内细胞标志物概况。(左)人类组织中不同细胞类型的细胞标志物数量统计;(右)小鼠组织中不同细胞类型的细胞标志物数量统计。
基于进化基因组和功能基因组数据的灵长类特异新基因数据库—GenTree
灵长类特异蛋白编码基因可推动表型演化,但目前只有少量功能研究。原因之一是缺乏相对可靠的灵长类特异基因(primate-specific genes or PSGs)数据集。由于基因年龄推断方法的差别以及新基因注释质量低等原因,已发表的PSG数据集间存在较大差别。中国科学院动物研究所张勇团队联合合作伙伴通过整合进化基因组和功能基因组数据开发了PSG数据库GenTree。GenTree可用来分析基因何时起源、如何起源及其功能。该工作进一步评估了常用的年龄推断方法及基因注释方法的优缺点后,鉴定了846个PSGs(含192个人特异的新基因),并发现PSG倾向于参与精子发生、免疫反应、母胎互作及胎脑发育等快速演化的生命过程。总而言之,该工作开发了一个专门的新基因数据库,产生了相对高质量的PSG列表并推测了其功能。年龄推断方法、基因注释方法的评估及新基因的功能特点对研究其它物种种系特异基因的工作具有普遍参考意义。
该成果发表于《基因组研究》期刊。
数据库链接:
http://gentree.ioz.ac.cn/
原文信息:
Shao Y, Chen C, Shen H, He BZ, Yu D, Jiang S, et al. GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res 2019;29:682–96. PMID: 30862647.
原文链接:
https://doi.org/10.1101/gr.238733.118
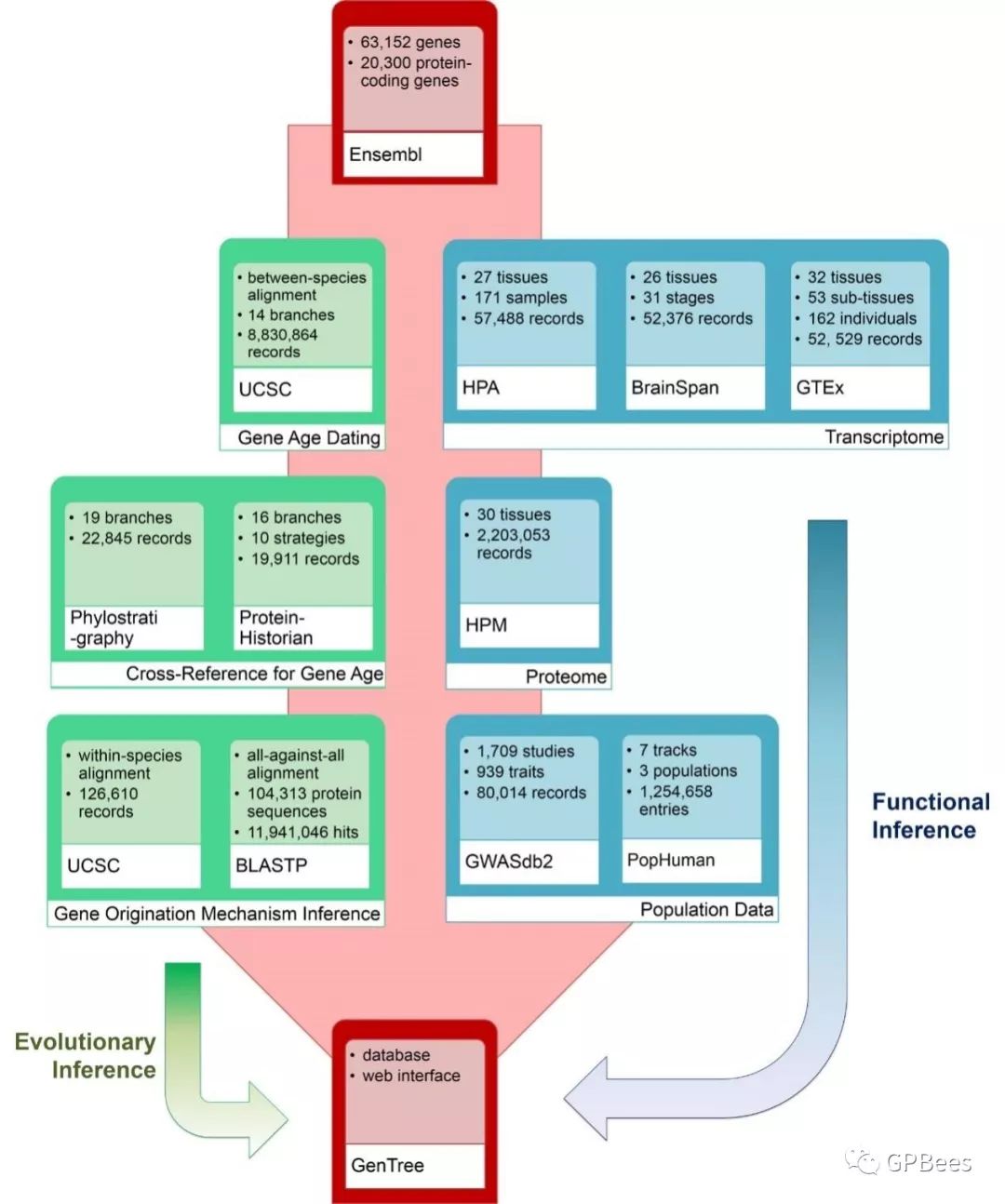
图:GenTree数据总览。(红色)Ensembl提供的基因注释等基本信息;(绿色)基于基因组共线性的基因年龄推断、其他研究的年龄推断佐证及起源机制推断等演化推断信息;(蓝色)包括转录组数据(HPA数据、GTEx数据、BrainSpan数据)、蛋白组数据(HPM数据)和群体数据(全基因组关联分析数据、群体遗传学数据)在内的基因功能相关信息。
整合Hi-C和FISH重构三维基因组结构新方法—GEM-FISH
随着三维基因组构象捕获实验技术(3C)尤其是与通量测序相结合技术(Hi-C)的发展,解析三维基因组结构成为了研究基因调控的常用手段。精确的三维基因组结构重构对于研究基因调控等生物过程和功能具有非常重要的意义。目前,绝大多数三维基因组结构重构算法均只是基于Hi-C数据。清华大学曾坚阳研究团队将Hi-C和荧光原位杂交(FISH)数据整合起来,基于流形学习算法框架(GEM)提出了新的三维基因组结构建模方法—GEM-FISH。该方法采用一种分治策略,首先利用Hi-C和FISH数据构建拓扑关联结构域(TAD)之间的相对结构,再利用Hi-C数据构建TAD内部结构,最后将两者结合起来根据大分子能量性质优化微调,获得最终的结构。与现有方法相比,这一方法重构的基因组结构更为精确,平均相对误差更小,并且能够准确揭示出活跃状态和失活状态X染色体的差异。该方法被用来分析子结构分隔区在染色体三维空间的分布并得到了FISH实验数据的进一步验证。另外,该方法还可以用来分析超级增强子在染色体三维空间的分布,为基因调控研究提供更多的线索。
该成果发表于《自然通讯》期刊。
工具链接:
https://github.com/ahmedabbas81/GEM-FISH
原文信息:
Abbas A, He X, Niu J, Zhou B, Zhu G, Ma T, et al. Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes. Nat Commun 2019;10:2049. PMID: 31053705.
原文链接:
https://doi.org/10.1038/s41467-019-10005-6
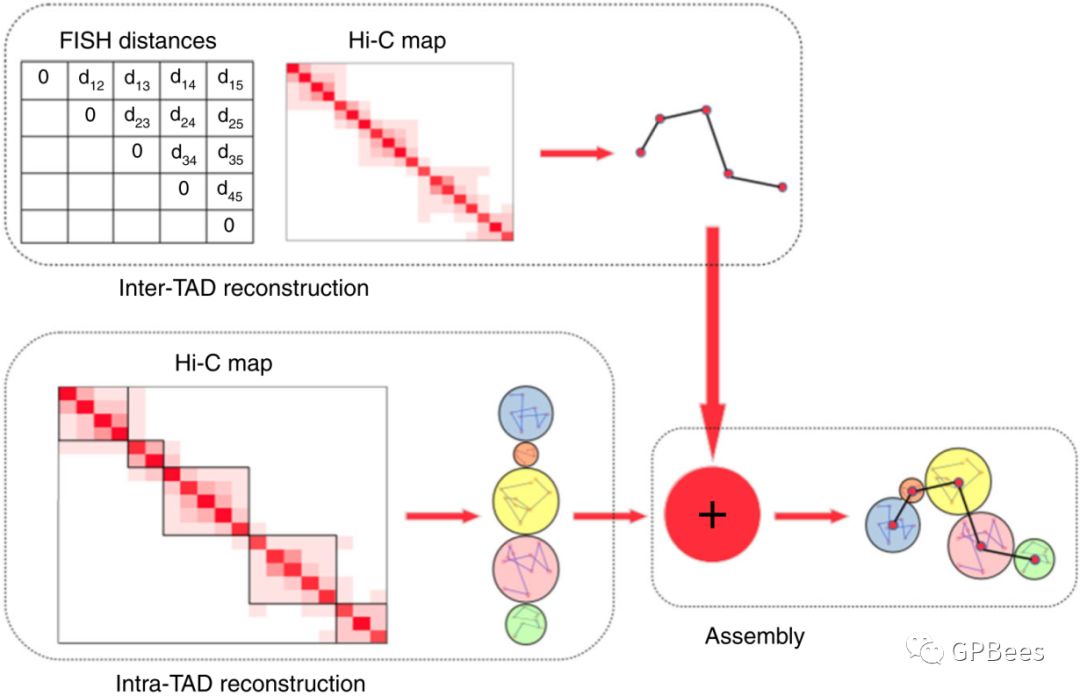
图:GEM-FISH通过整合Hi-C和FISH数据采用分治策略建模过程概览。(上)利用Hi-C和FISH数据以及生物物理的先验知识构建TAD水平的三维染色体结构;(左下)利用Hi-C数据和生物物理的先验知识构建染色体TAD内部的三维结构;(右下)将染色体TAD水平的结构与TAD内部的结构相结合,进一步调整生成最终完整的三维染色体结构。
细胞动态行为推断算法助力肝细胞癌免疫微环境的解析
单细胞转录组测序技术为系统解析肿瘤微环境的组成与特征提供了强大的实验工具,但如何推断肿瘤微环境中免疫细胞的动态行为是亟需解决的生物信息学问题。北京大学张泽民、任仙文团队,北京世纪坛医院彭吉润团队和勃林格殷格翰药业公司刘康团队以肝细胞癌为例进行攻关,通过系统整合当前细胞动态行为推断算法并建立配套的统计检验方法,揭示了肝癌病人腹水免疫细胞的来源和肿瘤树突状细胞的动态行为。通过集成基于基因表达谱相似性推断算法的高普适性、基于线粒体突变推断算法的高可信度和RNA剪接动力学分析可指示细胞动态行为的方向等不同算法的优点,并进行假设检验,揭示了肝癌病人腹水淋巴系免疫细胞主要来源于外周血,而髓系免疫细胞主要来源于肿瘤及癌旁组织。肿瘤中的一群LAMP3+树突状细胞则具有从肿瘤向淋巴结迁移的能力,并可通过表达多种配体与受体调节T淋巴细胞的浸润与表型。这些发现对于理解肝癌的免疫逃逸机制和开发新的免疫治疗方法具有重要意义。
该成果发表于《细胞》期刊。
数据链接:
https://bigd.big.ac.cn/gsa-human/browse/HRA000069
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140228
原文信息:
Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamicsof single immune cells in hepatocellular carcinoma. Cell 2019;179:829–45.e20. PMID: 31675496.
原文链接:
https://doi.org/10.1016/j.cell.2019.10.003

图:利用单细胞测序技术与分析方法刻画肝癌动态免疫图谱。(A) 技术流程:结合不同单细胞测序技术对肝癌多个免疫相关组织进行测序与分析;(B) 结果概述:肝癌不同免疫相关组织呈现出细胞富集的特异性,且不同组织特别是肝癌组织、肝周淋巴结、腹水之间存在复杂的细胞迁移动态关系。
小鼠三胚层谱系时空转录组图谱—eGastrulation
细胞的空间位置信息以及细胞在组织中原位的状态具有十分重要的生物学价值。中国科学院生物化学与细胞生物学研究所景乃禾团队、中国科学院-马普学会计算生物学伙伴研究所/北京大学韩敬东团队和中国科学院广州生物医药与健康研究院/广州再生医学与健康广东省实验室彭广敦团队合作利用空间转录组测序技术,从时空动态的四维角度来研究小鼠早期胚胎原肠运动期间的细胞谱系,构建了胚胎着床后从多能干细胞退出到外、中、内三胚层建立的谱系发生关系树,在国际上首次获得了具有极高分辨率和完整度的时空体内细胞发育和命运图谱,全景式地展现了干细胞命运决定的分子表达谱。该研究建立了全基因组的时空表达数据库eGastrulation,供领域内研究者查询超过2万个基因的三维空间表达模式,分析共表达关系,并可实现单细胞的空间定位(zip code mapping),以及基于特征表达模式的基因模式分析。该数据库为目前国际上关于小鼠原肠运动时期最为全面和完整的交互性时空转录组数据库。
该成果发表于《自然》期刊。
数据库链接:
http://egastrulation.sibcb.ac.cn/
原文信息:
Peng G, Suo S, Cui G, Yu F, Wang R, Chen J, et al. Molecular architecture of lineage allocationand tissue organization in early mouse embryo. Nature 2019;572:528–32. PMID: 31391582.
原文链接:
https://doi.org/10.1038/s41586-019-1469-8
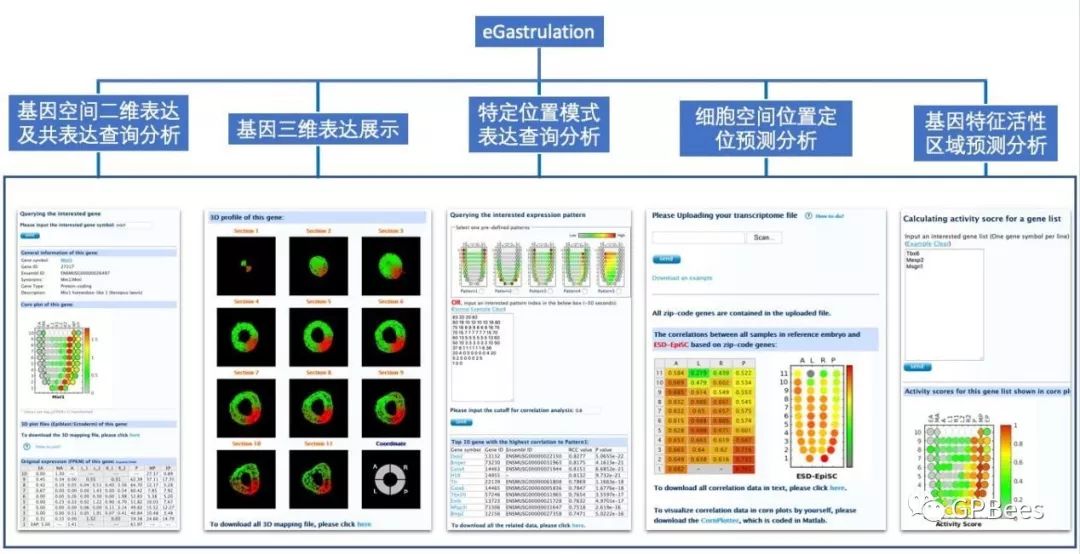
图:原肠运动时空三维数据库(eGastrulation)的功能实现
现存和古人族群的全基因组单核苷酸变异数据库—PGG.SNV
在医学遗传学领域的实践中,人们往往通过对突变位点的频率进行比较分析来筛选并判别突变是否可能致病。其潜在假设是孟德尔疾病相关的致病突变在自然人群中是稀有的。孟德尔疾病突变在不同族群中频率存在差别,因而用西方族群的基因组来研究或预测其他族群的突变功能和疾病风险可能会导致误判。中国科学院-马普学会计算生物学伙伴研究所徐书华团队构建了更具亚洲人群特色的全基因组单核苷酸变异数据库—PGG.SNV。PGG.SNV收录的基因组数据涵盖了800多个现存人类族群和来源于古DNA研究的100多个已消亡的人类族群,总共超过20万个基因组,因而在代表性人群数量和样本量上均超过目前被广泛使用的由西方学者主导的gnomAD数据库。PGG.SNV更显著的科学价值在于提供了人群、个体、基因和变异多个层面的种群遗传多样性和进化参数的估计,有助于更深入地解析人类基因组变异的功能和表型效应,理解其进化和医学意义。PGG.SNV数据库同时开通了配套的微信公众号“PGGbase”,方便微信用户通过智能手机查询。
该成果发表于《基因组生物学》期刊。
数据库链接:
https://www.pggsnv.org
原文信息:
Zhang C, Gao Y, Ning Z, Lu Y, Zhang X, Liu J, et al. PGG.SNV: understanding the evolutionary and medical implications of human single nucleotide variations in diverse populations. Genome Biol 2019;20:215. PMID: 31640808.
原文链接:
https://doi.org/10.1186/s13059-019-1838-5
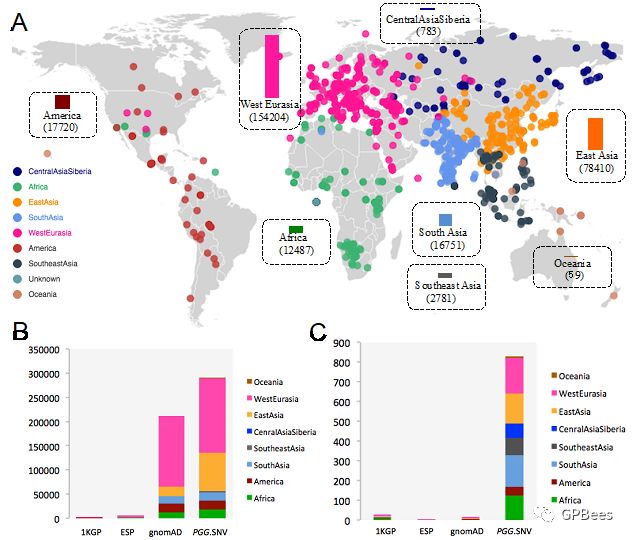
图:PGG.SNV基因组数据的人群和地理分布概览。(A)PGG.SNV数据库所覆盖人类族群的分布和基因组数量统计;(B)PGG.SNV与gnomAD等数据集在基因组数目上的比较;(C)PGG.SNV与gnomAD等数据集在族群数量上的比较。
利用人工智能算法分析单细胞ATAC-seq数据—SCALE
染色质开放区域是基因组编码生命信息的窗口,其中包含了各种各样重要的转录因子结合位点和其他类型的基因表达调控元件。单细胞ATAC-seq技术可以在单细胞水平上描绘染色质的开放图谱从而揭示细胞间在基因表达调控上的差异。然而,对于每一种真核细胞,其所有可能的染色质开放位点数目通常有几十万之多,这造成所谓的“维度灾难”;同时由于生物和技术的原因,许多潜在的开放区域没有信号、数据异常稀疏。因此,目前尚缺乏有效的方法来分析挖掘海量的单细胞ATAC-seq数据中宝贵的生物学信息。清华大学张强锋团队通过深度学习的方法,结合高斯混合模型和变分自编码器,提取数据低维的隐层特征,对单细胞ATAC-seq数据进行聚类、可视化、缺失值填补、降噪以及下游生物信息挖掘,有效地解决了数据高维度稀疏性的问题。该方法为解码单细胞表观遗传学提供了一个综合高效强大的工具,将有望在肿瘤、免疫、发育等领域的研究中得到广泛应用。
该成果发表于《自然通讯》期刊。
工具链接:
https://github.com/jsxlei/SCALE
原文信息:
Xiong L, Xu K, Tian K, Shao Y, Tang L, Gao G, et al. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat Commun 2019;10:4576. PMID:31594952.
原文链接:
https://doi.org/10.1038/s41467-019-12630-7
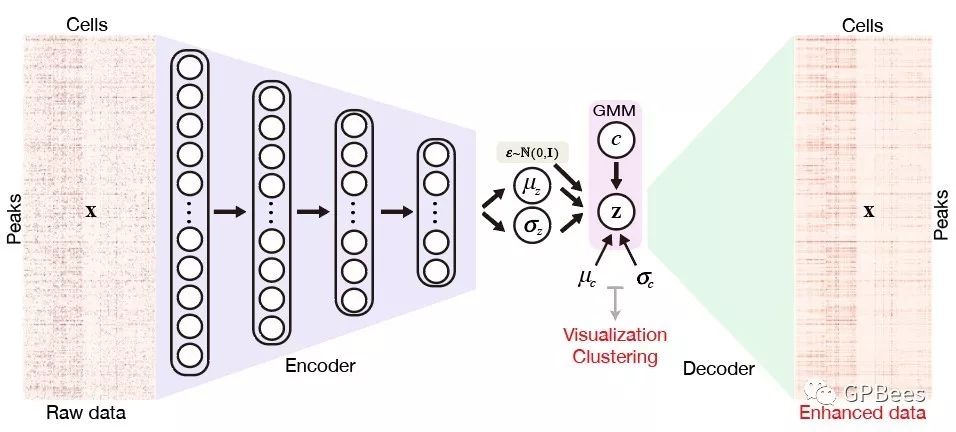
图: SCALE模型概览。SCALE采用一个编码器(encoder)和解码器(decoder)组成的变分自编码器(VAE)框架,输入是peak×cell矩阵表示的单细胞数据(raw data),输出是提取的可以用于可视化(visualization)和聚类(clustering)的隐层特征z,以及消除了噪音并填补了缺失值的增强单细胞数据(enhanced data)。SCALE编码器是一个四层的神经网络,解码器是一个隐层特征z和增强数据直连的单层神经网络,隐层特征在流形空间被μ_c和σ_c^ 参数化的高斯混合模型所约束。
中国人群全基因组测序研究及北方汉族参考基因组建立
在人群中进行规模化的个体基因组序列与其临床表型的整合分析研究是精准医学的重要方向,将极大地促进探索和揭示个体从健康到疾病的发生发展过程中的相关分子机制及早期信号。面向我国精准医学研究的重大需求,2016年中国科学院北京基因组研究所牵头启动了中国科学院的中国人群精准医学研究计划(CASPMI),采用多平台结合的方法建立了高质量的中国北方汉族参比基因组NH1.0;同时根据前期对近600名参加个体的全基因组深度测序,建立了中国人群的全基因组遗传变异图谱,鉴定了中国人群(东亚人群)特异性高频多态位点。结合基因型-表型关联分析,发现位于编码组蛋白乙酰化转移酶基因KAT8的SNP位点rs1549293与男性腰围显著相关;通过中国南北方人群的遗传差异位点分析,提出叶酸代谢相关基因MHTFR上影响酶活的多态位点rs1801133的T等位基因(677T)在北纬35–45度之间存在一个高频区。这些研究成果将为中国精准医学研究提供重要支持。
该成果发表于《基因组蛋白质组与生物信息学报》期刊。
数据链接:
https://bigd.big.ac.cn/gsa/browse/CRA000631
https://bigd.big.ac.cn/gwh/Assembly/19/show
原文信息:
Du Z, Ma L, Qu H, Chen W, Zhang B, Lu X, et al. Whole genome analyses of Chinese population and de novo assembly of a Northern Han genome. Genomics Proteomics Bioinformatics 2019;17:229–47. PMID: 31494266.
原文链接:
https://doi.org/10.1016/j.gpb.2019.07.002
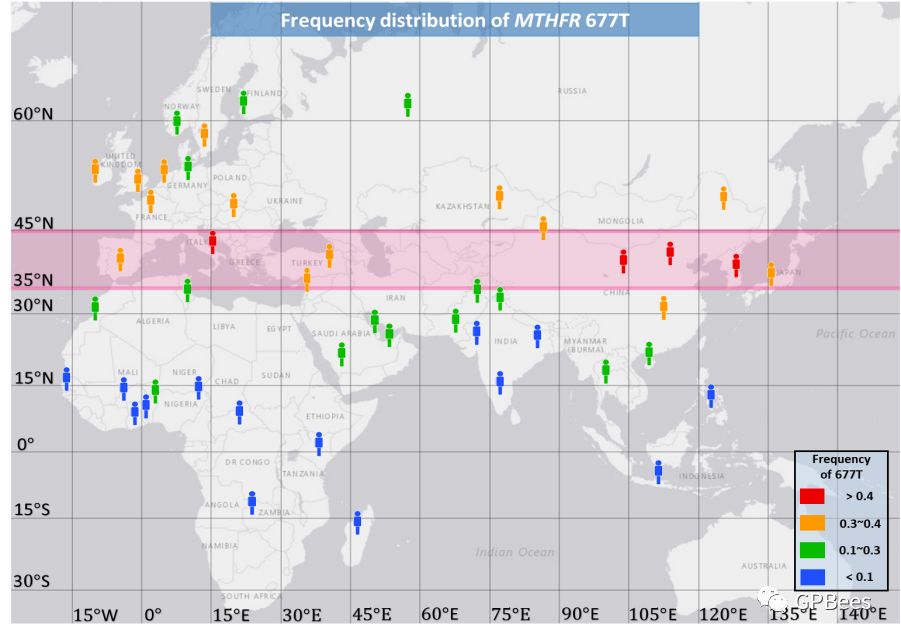
图:MHTFR多态位点rs1801133的T等位基因(677T)全球范围在北纬35–45度之间存在一个高频区带。
人体肠道可培养细菌参考基因组数据集及活体菌株库—CGR
高质量的参考基因组和活体菌株是深入研究疾病与肠道菌群相互作用机制的基础。为了完善并进一步扩充现有肠道菌株库和参考基因组数据集,深圳华大生命科学研究院肖亮、贾慧珏、李俊桦团队利用培养组学方法分离了超过6000株来自健康人体粪便样本的肠道细菌菌株,以其基因组数据为基础构建了1520株高质量的肠道细菌基因组数据集—Culturable Genome Reference (CGR),发现了338个物种分类群,其中超过三分之一是新的细菌物种。这项研究极大丰富了现有肠道微生物物种的多样性,将肠道微生物宏基因组分析、基因组SNP分析、功能分析和重要肠道菌的泛基因组分析提升到新维度,加深了人们对于人体肠道微生物的认知。这是首次通过大规模培养的技术手段获得如此多数量的活体菌株及相应的高质量细菌基因组数据。这一成果对于实现在菌株层面精准解析肠道微生物与疾病之间的关系具有重要的科研价值,也为微生物组的临床应用转化提供了宝贵的菌株资源支持。
该成果发表于《自然生物技术》期刊。
数据库链接:
https://db.cngb.org/cnsa/project/CNP0000126/public/
原文信息:
Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat Biotechnol 2019;37:179–85. PMID: 30718868.
原文链接:
https://doi.org/10.1038/s41587-018-0008-8
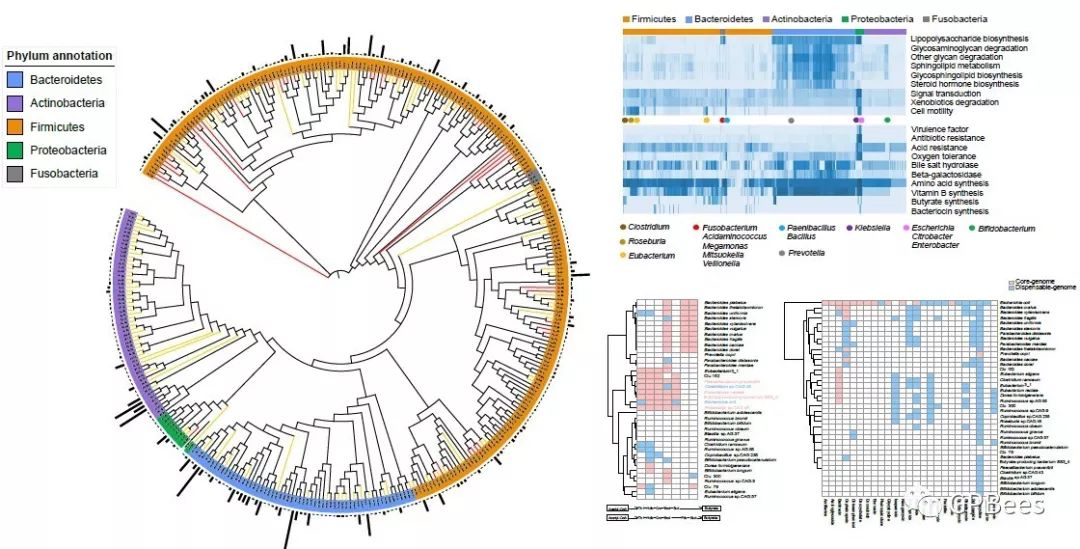
图:肠道可培养细菌参考基因组(CGR)系统进化与功能分布概览。(左)基于CGR 中1520株肠道细菌全基因组数据的系统进化树;(右上)CGR 中1520株肠道细菌功能(基于KEGG数据库注释结果)分布图谱;(右下)CGR中38种代表性菌株泛基因组分析以及丁酸和抗生素抗性功能通路分布。