


ADMIN | 2023-03-02 23:18:10
评审委员会
2023年3月3日

数据驱动的蛋白质从头设计
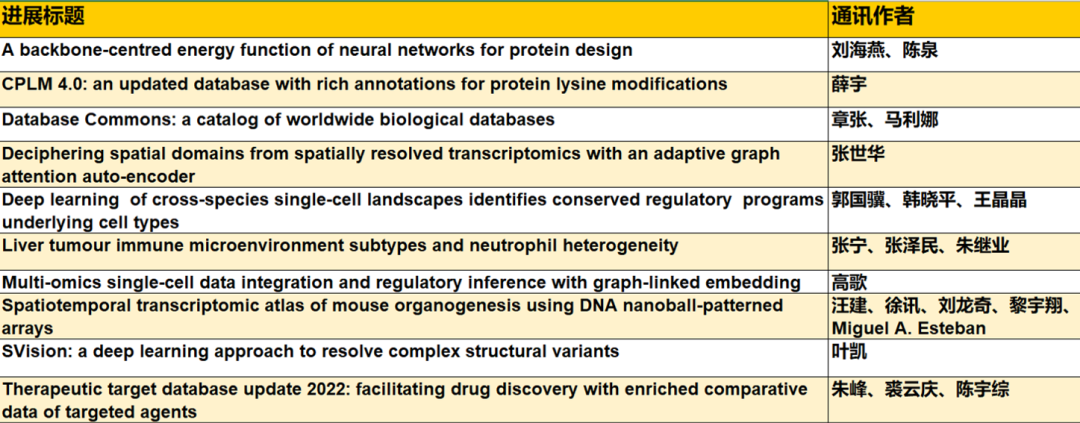
蛋白质从头设计可以打破自然进化的约束,按需设计自然界不存在的具有特定空间结构和预期功能的蛋白质,在生物医药、绿色制造等领域具有广阔的应用前景。中国科学技术大学刘海燕、陈泉团队基于数据驱动原理建立用人工神经网络表示的统计能量模型,首次实现并实验验证能够在氨基酸序列待定的前提下连续广泛地搜索主链结构空间,自动产生有高可设计性的主链结构,通过此设计路线成功设计多个自然界不存在的全新结构的人工蛋白,建立了一条全新的蛋白设计路线。该工作实现了关键核心技术的原始创新,为按需设计功能蛋白奠定了坚实的基础。
该成果发表于Nature
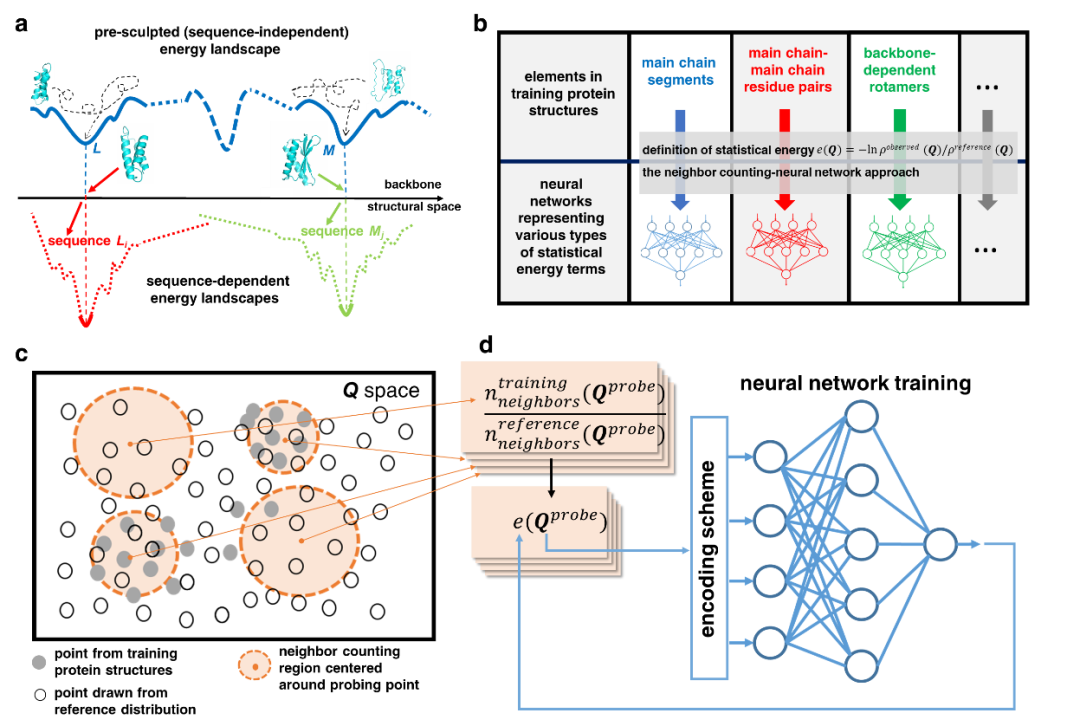
图:用SCUBA模型进行蛋白质设计的原理
数据链接
https://www.wwpdb.org/pdb?id=pdb_00007dmf
https://www.wwpdb.org/pdb?id=pdb_00007dkk
https://www.wwpdb.org/pdb?id=pdb_00007dko
https://www.wwpdb.org/pdb?id=pdb_00007dgu
https://www.wwpdb.org/pdb?id=pdb_00007dgw
https://www.wwpdb.org/pdb?id=pdb_00007dgy
https://www.wwpdb.org/pdb?id=pdb_00007fbb
https://www.wwpdb.org/pdb?id=pdb_00007fbc
https://www.wwpdb.org/pdb?id=pdb_00007fbd
工具链接
https://doi.org/10.5281/zenodo.4533424
原文信息
Huang B, Xu Y, Hu X, Liu Y, Liao S, Zhang J, et al. A backbone-centred energy function of neural networks for protein design. Nature 2022;602:523–8. PMID: 35140398.
原文链接
https://www.nature.com/articles/s41586-021-04383-5

▲ 长按阅读原文
丰富注释的赖氨酸修饰数据库——CPLM 4.0
蛋白质赖氨酸修饰是重要的蛋白质翻译后修饰之一,它通过修饰基团共价偶联到底物蛋白的特定赖氨酸位点上,进而影响生物体内的多种生理病理过程。华中科技大学薛宇团队构建的第四版赖氨酸修饰数据库中,整理了文献及其他8个翻译后修饰位点数据库中的数据,将收录的赖氨酸修饰信息条目拓展至592,606条,其中包含新收录的9种重要赖氨酸修饰类型。至此,CPLM 4.0 整合了219个物种中的29种赖氨酸修饰,共包含463,156个鉴定的赖氨酸修饰位点。此外,团队利用105个公共数据库为这些收录的赖氨酸修饰蛋白质作了详细的注释,为研究者提供了全面整合的综合信息。
该成果发表于Nucleic Acids Research
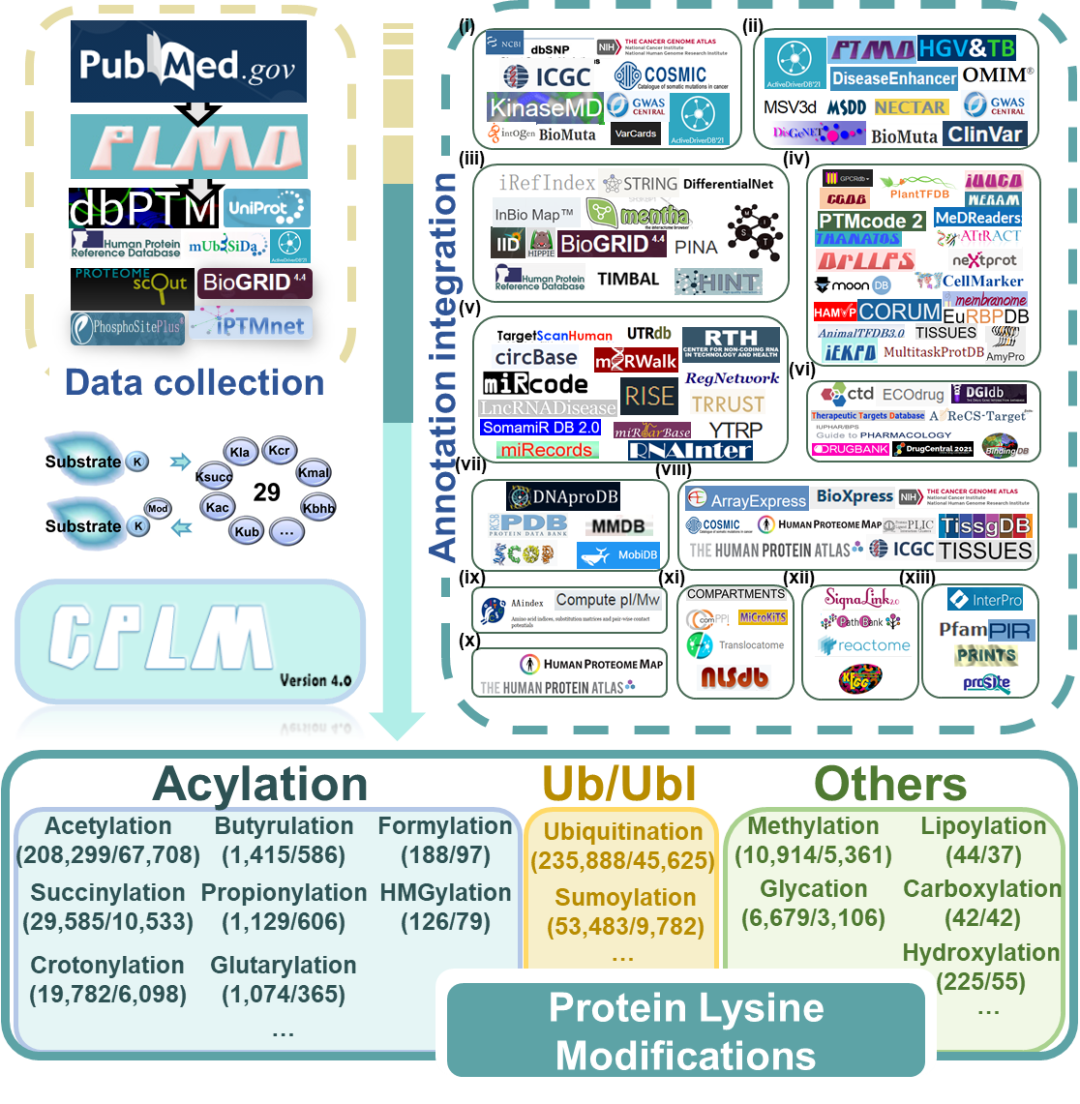
图:CPLM 4.0 收录数据
数据库链接
https://cplm.biocuckoo.cn/
原文信息
Zhang W, Tan X, Lin S, Gou Y, Han C, Zhang C, et al. CPLM 4.0: an updated database with rich annotations for protein lysine modifications. Nucleic Acids Research 2022;50:D451–9. PMID: 34581824.
原文链接
https://academic.oup.com/nar/article/50/D1/D451/6377396

▲ 长按阅读原文
全球生物数据库目录——Database Commons
生物数据库作为全球各类生命科学研究的基础支撑,极大促进了大数据向知识的转化,并推动了众多研究领域的重要创新。为建立全球生物数据库目录,中国科学院北京基因组研究所(国家生物信息中心)章张、马利娜团队构建了Database Commons数据库。该团队联合国内外多家科研机构,历经7年多的时间开展数据积累和功能完善,基于8992篇文献,审编了分布于72个国家/地区的由1992个机构开发的5899个生物数据库。同时,设计了z-index用于评估数据库的科学影响,并根据数据库文章引用和z-index对所有生物数据库及其隶属机构和国家进行排名。因此,Database Commons提供了全球生物数据库的系列统计数据和趋势,为更好地了解数据库发展态势及其对生命健康科学的影响提供全球视角。
该成果发表于Genomics, Proteomics & Bioinformatics
图:Database Commons数据库内容概要(统计信息截至2023年2月28日)
数据库链接
https://ngdc.cncb.ac.cn/databasecommons/
原文信息
Ma L, Zou D, Liu L, Shireen H, Abbasi AA, Bateman A, et al. Database Commons: a catalog of worldwide biological databases. Genomics, Proteomics & Bioinformatics 2022;DOI: 10.1016/j.gpb.2022.12.004. PMID: 36572336.
原文链接
https://www.sciencedirect.com/science/article/pii/S1672022922001693?via%3Dihub

▲ 长按阅读原文
基于空间转录组的生物组织亚结构解析新工具——STAGATE
空间转录组学技术的重大进展使研究人员得以在全基因组层面测量组织切片中特定空间位点的基因表达信息。精确破译空间域是空间转录组数据解析中最基本和关键的环节。然而,现有方法并没有充分利用数据的空间位置信息,导致识别结果极易受到技术噪音的影响。中国科学院数学与系统科学研究院张世华团队针对不同空间转录组技术、不同生物组织建立了破译生物组织空间亚结构的人工智能算法与工具——STAGATE。随着空间转录组技术的日益进步、推广和数据的不断积累,该方法为解析空间数据提供了一个综合高效的工具,将对大规模空间转录组数据的精确解析提供助力。
该成果发表于Nature Communications
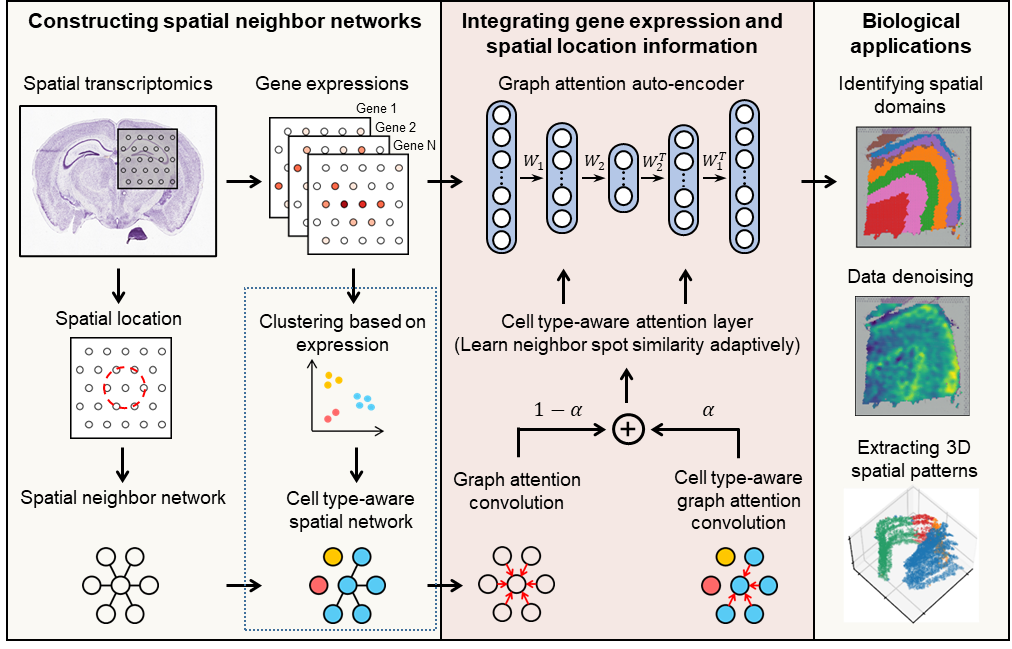
图:STAGATE算法的工作原理示意图
工具链接
https://github.com/zhanglabtools/STAGATE
https://doi.org/10.5281/zenodo.6330702
原文信息
Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nature Communications 2022;13:1739. PMID:35365632.
原文链接
https://www.nature.com/articles/s41467-022-29439-6

▲ 长按阅读原文
基于人工智能神经网络的基因组解读系统——Nvwa
预测基因表达和解析基因调控机制一直是基因组学的重要目标。浙江大学郭国骥、韩晓平和王晶晶团队利用自主构建的高通量单细胞测序平台Microwell-seq绘制了斑马鱼、果蝇和蚯蚓的全身单细胞转录组图谱,并探究了八种代表性后生动物细胞类型的跨物种可比性。团队进一步提出了深度学习模型Nvwa(女娲),首次完全基于基因组序列实现了单细胞分辨率下的基因表达预测,学习了谱系特异性调控基序,并解析了各组织细胞类型的调节程序。团队基于Nvwa模型Filter的跨物种比较,发现同源Filter倾向于保持细胞类型的特异性。该工作首次建立了物种层面基因组编码细胞图谱的整合模型,并为解码多物种基因调控程序和预测元件突变表型提供了宝贵资源。
该成果发表于Nature Genetics
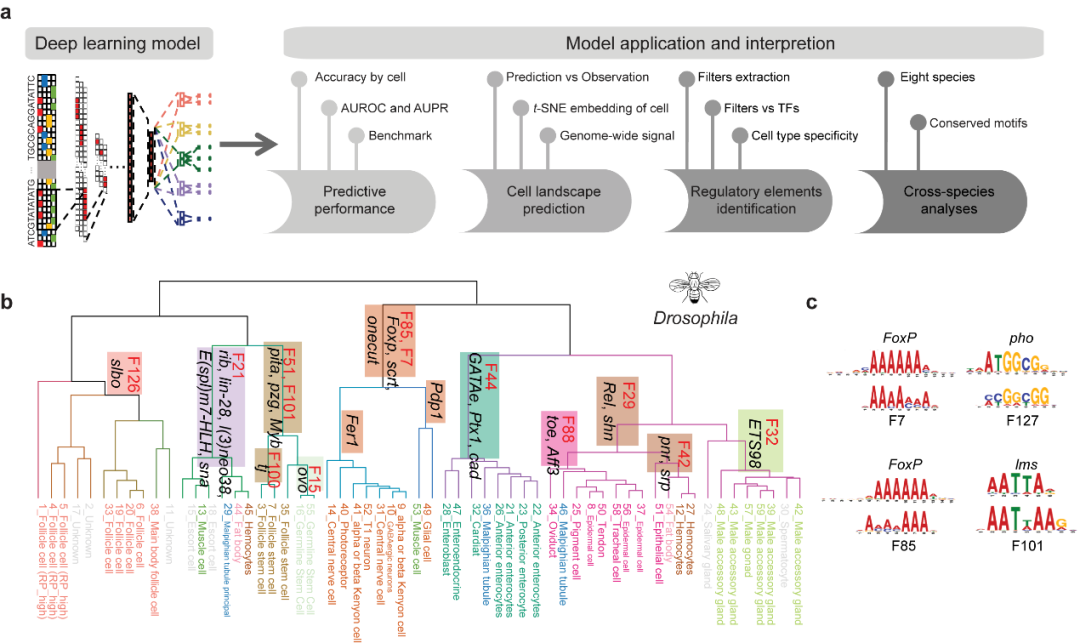
图:深度学习模型Nvwa(女娲)
数据链接
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178151
https://figshare.com/s/ecc05b1051fb5678fd3e
http://bis.zju.edu.cn/nvwa
工具链接
https://github.com/JiaqiLiZju/Nvwa/
https://zenodo.org/record/6806748
原文信息
Li J, Wang J, Zhang P, Wang R, Mei Y, Sun Z, et al. Deep learning of cross-species single-cell landscapes identifies conserved regulatory programs underlying cell types. Nature Genetics 2022;54:1711–20. PMID: 36229673.
原文链接
https://www.nature.com/articles/s41588-022-01197-7

▲ 长按阅读原文
肝癌免疫微环境亚型和中性粒细胞异质性
免疫微环境异质性是肿瘤耐药和转移的重要原因之一;系统探究免疫微环境的异质性规律对治疗选择、疗效预测及新靶点开发意义重大。北京大学张宁、张泽民和朱继业团队合作,综合利用单细胞测序、空间转录组测序和外显子测序对189个人和小鼠肝癌样本的微环境进行分析。该研究首次在单细胞精度刻画了肝癌的五种免疫微环境亚型并命名为TIMELASER分型系统,揭示其细胞组成、空间分布、驱动突变、趋化因子网络和预后相关性。该研究也首次揭示肝癌的中性粒细胞异质性,阐明CCL4阳性和PD-L1阳性的肿瘤相关中性粒细胞的促肿瘤机制,最终通过构建新的小鼠自发肝癌动物模型证明靶向肿瘤相关中性粒细胞有望形成新的肝癌治疗策略。
该成果发表于Nature
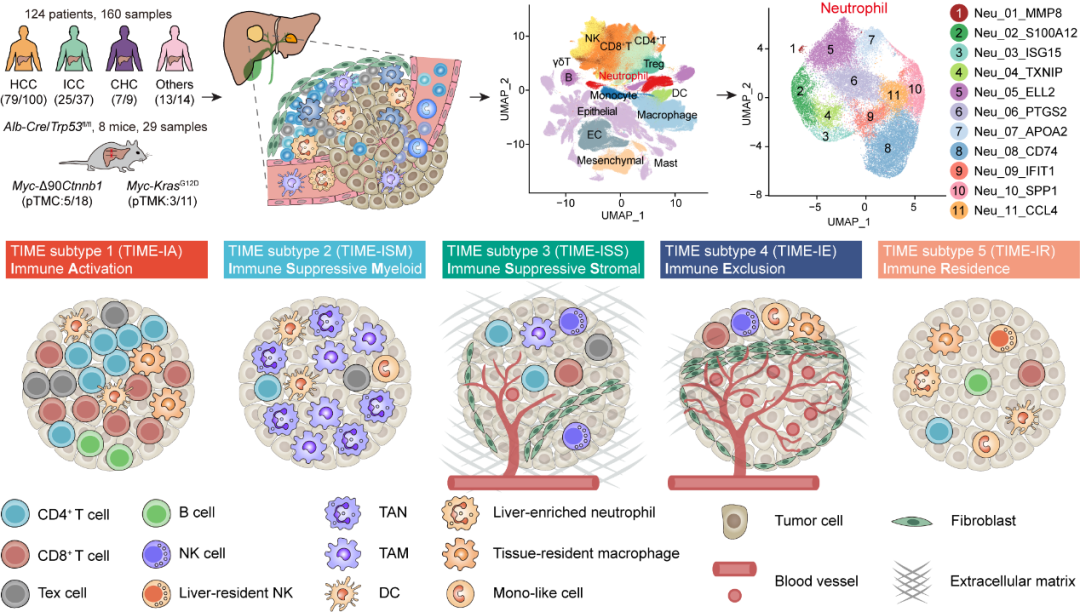
图:肝癌免疫微环境TIMELASER分型系统和中性粒细胞异质性
数据链接
https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA007744
工具链接
http://meta-cancer.cn:3838/scPLC/
https://github.com/meta-cancer/scPLC
原文信息
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022;612:141–7. PMID: 36352227.
原文链接
https://www.nature.com/articles/s41586-022-05400-x

▲ 长按阅读原文
单细胞多组学数据整合与调控推断算法——GLUE
单细胞多组学研究对于精确解析细胞状态与基因调控机制具有重要意义,但由于不同组学特征空间不同、异质性强,单细胞多组学数据的整合分析面临挑战。为解决这一问题,北京大学/昌平实验室高歌团队提出基于生成-对抗学习的图耦联策略,通过直接对调控关系进行建模以实现跨组学特征在隐空间的自适应关联。以此为基础开发的GLUE算法首次实现了统一框架下百万级非配对单细胞多组学数据的无监督整合与同步调控推断,可广泛应用于包括基因表达、染色质开放性、DNA甲基化等多种组学数据的分析挖掘。
该成果发表于Nature Biotechnology
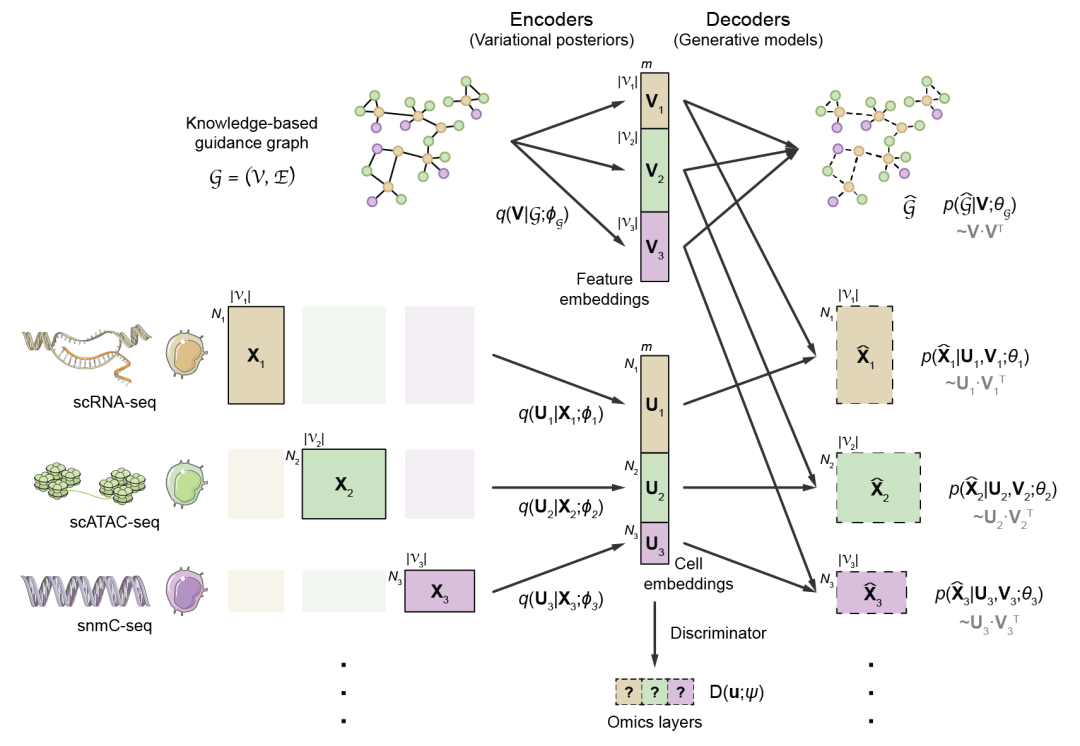
图:GLUE模型的结构示意图
工具链接
https://github.com/gao-lab/GLUE
原文信息
Cao ZJ, Gao G. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nature Biotechnology 2022;40:1458–66. PMID: 35501393.
原文链接
https://www.nature.com/articles/s41587-022-01284-4

▲ 长按阅读原文
Stereo-seq绘制高精度生命全景时空基因表达地图
细胞是生命的基本功能单元。细胞类型、定位和细胞间通讯的分析对于理解器官功能、个体发育、人类疾病和物种器官演化至关重要。华大生命科学研究院汪建、徐讯主导的团队联合中国科学院广州生物医药与健康研究院、郑州大学、深圳湾实验室、广东省基因组读写实验室等多家机构基于自主DNA纳米球测序技术,研发了高精度大视场空间转录组技术Stereo-seq,将认识生命的分辨率推进到了500 nm的亚细胞级。相比过去同类技术,分辨率提升了200倍,视野大小提升了483倍。基于该技术,华大首次绘制了小鼠胚胎发育过程中迄今为止最高精度最全面的时空基因表达数据集,在全胚胎尺度分析了细胞类型的空间多样性,并配套开发了时空组学数据分析平台和在线交互数据库。在此基础上,华大主导发起了时空组学国际联盟(STOC),吸引了来自30多个国家200多个科研团队参与。
该成果发表于Cell
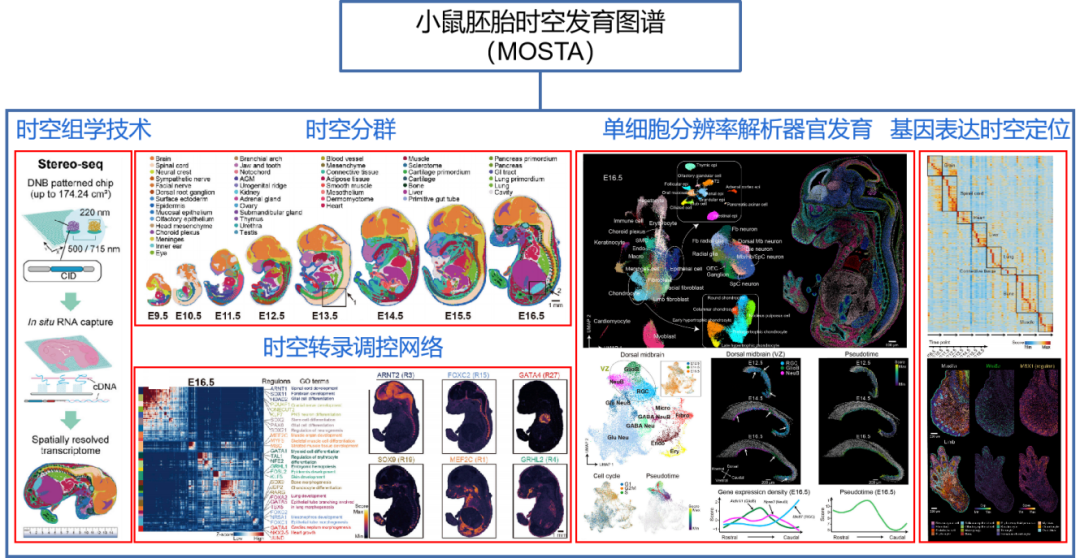
图:利用大视场纳米级分辨率时空组学技术构建具有单细胞分辨率的小鼠胚胎器官发育时空基因表达图谱
数据链接
https://db.cngb.org/search/project/CNP0001543
工具链接
https://github.com/BGIResearch/SAW
https://db.cngb.org/stomics/mosta/
原文信息
Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022;185:1777–92.e21. PMID:35512705.
原文链接
https://www.sciencedirect.com/science/article/pii/S0092867422003993

▲ 长按阅读原文
基因组复杂结构变异检测方法——SVision
基因组结构变异是物种进化和疾病发生的重要驱动因素,对结构变异的全面精准检测表征是基因组精细结构研究的核心环节,然而目前尚缺乏针对基因组重复区域复杂结构变异的检测策略。西安交通大学叶凯团队针对基因组重复区域背景噪声高、复杂结构变异类型未知且建模难,通过将基因组结构变异检测从序列空间转换为图像空间,实现了简单和复杂类型结构变异的高性能检测和准确表征。团队开发了基于深度学习的多目标识别方法SVision,该方法无需依赖先验信息,能够从长读长测序数据中自动检测和表征未知类型的基因组结构变异,为后续多种生物医学应用场景的全类型基因组结构变异研究提供了有力工具和新方案。
该成果发表于Nature Methods
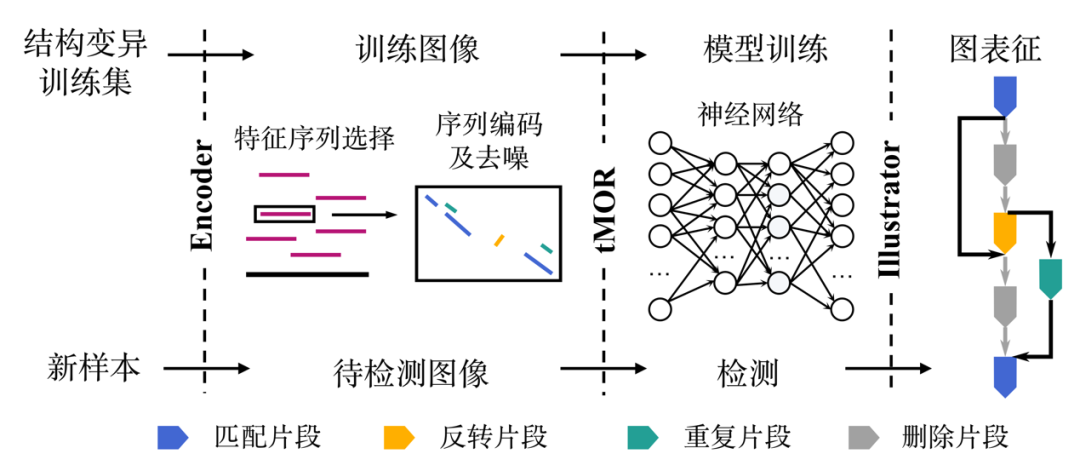
图:SVision算法流程图及三个主要模块
工具链接
https://github.com/xjtu-omics/SVision
https://doi.org/10.24433/CO.8937098.v1
原文信息
Lin J, Wang S, Audano PA, Meng D, Flores JI, Kosters W, et al. SVision: a deep learning approach to resolve complex structural variants. Nature Methods 2022;19:1230–3. PMID: 36109679.
原文链接
https://www.nature.com/articles/s41592-022-01609-w

▲ 长按阅读原文
疗效药物靶标的比较图谱构建及数据库开发——TTD
随着药物靶标(以下简称“药靶”)数据的不断累积,针对药靶数据的比较性研究(如对药靶活性分子的结构比较与分类、类药性比较与排序等)对新药发现至关重要。基于此,浙江大学朱峰、裘云庆团队和清华大学/宁波大学陈宇综团队合作开展了全面的药靶比较性研究,构建了药靶所有活性分子的“结构-活性”、“类药性”等比较图谱。研究涵盖了超过35,000个药物(包括FDA已批准、临床研究、临床前等药物)作用的疗效药靶,并将结果整合到团队前期开发的TTD数据库中。相关研究结果不仅可以提供药靶活性分子的骨架类型、定量构效关系和活性悬崖等重要信息,更有助于促进对药靶可药靶性的认识和对活性分子类药性的深入理解,为药物设计与新药发现提供了不可或缺的重要支持。
该成果发表于Nucleic Acids Research
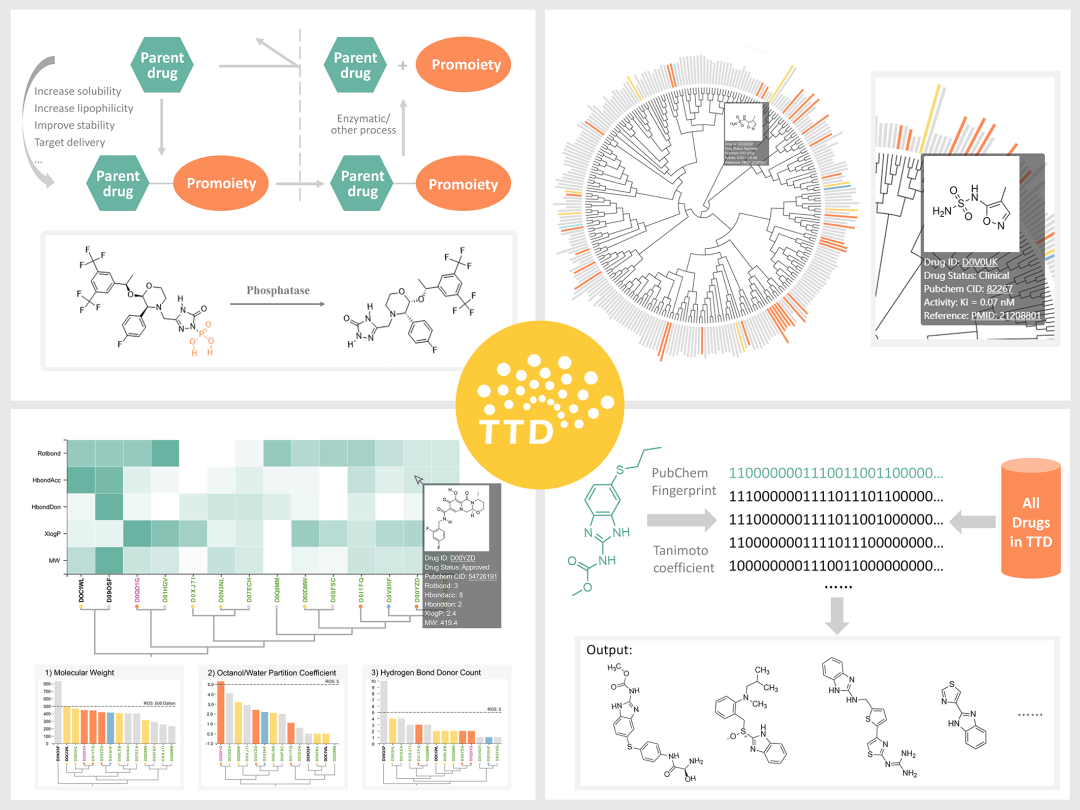
图:疗效药物靶标的比较图谱构建
数据库链接
http://bidd.group/group/cjttd/
原文信息
Zhou Y, Zhang Y, Lian X, Li F, Wang C, Zhu F, et al. Therapeutic target database 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Research 2022;50:1398–407. PMID: 34718717.
原文链接
https://academic.oup.com/nar/article/50/D1/D1398/6413598

▲ 长按阅读原文
About GPB

Genomics, Proteomics & Bioinformatics(基因组蛋白质组与生物信息学报,简称GPB)于2003年创刊,是由中国科学院主管、中国科学院北京基因组研究所(国家生物信息中心)与中国遗传学会共同主办的英文学术期刊,由Elsevier金色开放获取(Gold Open Access)出版。刊载来自世界范围内组学、生物信息学及相关领域的优质稿件。现为中国科学引文数据库(CSCD)和中国科技论文与引文数据库(CSTPCD)核心期刊,被SCIE、PubMed / MEDLINE、Scopus等数据库收录。2022年公布的官方数据显示,CiteScore为12.0,2年和5年Impact Factor分别为6.409和10.196。期刊由科技部等七部门联合实施的“中国科技期刊卓越行动计划”资助(2019–2023)。

(扫码访问期刊主页)